Orbital-resolved and k-resolved bonding descriptors#
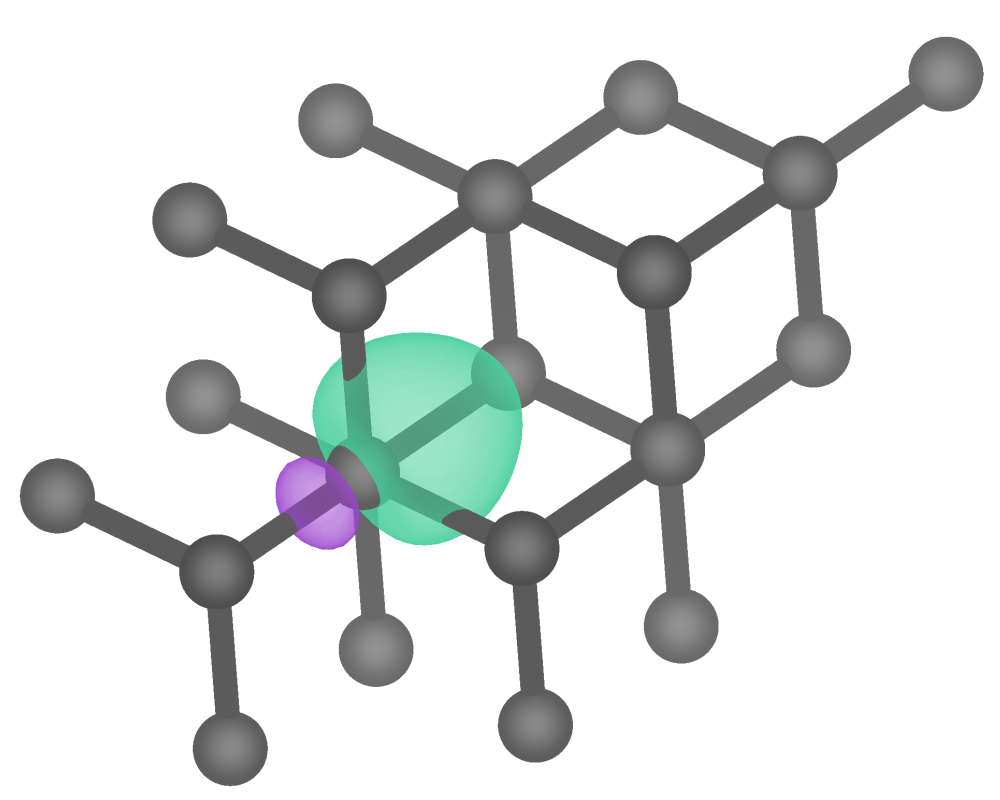
In the basics, we demonstrated the core functionality of pengwann by calculating, integrating and plotting the Wannier orbital Hamilton population (WOHP) and the Wannier orbital bond index (WOBI) for the C-C bond in diamond. We will now return to diamond once again to give a more detailed account of the outputs of pengwann, particularly with regards to resolving descriptors such as the WOHP with respect to axes other than the energy alone.
Orbital-resolved descriptors#
Manipulating Interaction objects#
from pengwann.geometry import Geometry, identify_interatomic_interactions
cutoffs = {('C', 'C'): 1.6}
geometry = Geometry.from_xyz(seedname='wannier90', path='inputs')
interactions = identify_interatomic_interactions(geometry, cutoffs)
print(interactions)
Atomic interactions
===================
C8 <=> C9
As introduced in the basics, the interaction between two atoms i and j is represented by an AtomicInteraction data structure:
print(interactions[8, 9])
Atomic interaction C8 <=> C9
============================
DOS matrix => Not calculated
WOHP => Not calculated
WOBI => Not calculated
IWOHP => Not calculated
IWOBI => Not calculated
Population => Not calculated
Charge => Not calculated
Associated Wannier interactions
-------------------------------
0[0, 0, 1] <=> 1[0, 0, 0]
0[0, 0, 1] <=> 3[0, 0, 0]
0[0, 0, 1] <=> 4[0, 0, 0]
0[0, 0, 1] <=> 7[0, 0, 0]
2[1, 0, 0] <=> 1[0, 0, 0]
2[1, 0, 0] <=> 3[0, 0, 0]
2[1, 0, 0] <=> 4[0, 0, 0]
2[1, 0, 0] <=> 7[0, 0, 0]
5[1, 1, 1] <=> 1[0, 0, 0]
5[1, 1, 1] <=> 3[0, 0, 0]
5[1, 1, 1] <=> 4[0, 0, 0]
5[1, 1, 1] <=> 7[0, 0, 0]
6[0, 1, 0] <=> 1[0, 0, 0]
6[0, 1, 0] <=> 3[0, 0, 0]
6[0, 1, 0] <=> 4[0, 0, 0]
6[0, 1, 0] <=> 7[0, 0, 0]
Previously, we concerned ourselves only with the total WOHP and WOBI for the interaction between C8 and C9, but we can also resolve this interaction with respect to the interactions between individual Wannier functions:
for w_interaction in interactions[8, 9]:
print(w_interaction)
Wannier interaction 0[0, 0, 1] <=> 1[0, 0, 0]
=============================================
DOS matrix => Not calculated
H_ij => Not calculated
P_ij => Not calculated
IWOHP => Not calculated
IWOBI => Not calculated
Population => Not calculated
Wannier interaction 0[0, 0, 1] <=> 3[0, 0, 0]
=============================================
DOS matrix => Not calculated
H_ij => Not calculated
P_ij => Not calculated
IWOHP => Not calculated
IWOBI => Not calculated
Population => Not calculated
Wannier interaction 0[0, 0, 1] <=> 4[0, 0, 0]
=============================================
DOS matrix => Not calculated
H_ij => Not calculated
P_ij => Not calculated
IWOHP => Not calculated
IWOBI => Not calculated
Population => Not calculated
Wannier interaction 0[0, 0, 1] <=> 7[0, 0, 0]
=============================================
DOS matrix => Not calculated
H_ij => Not calculated
P_ij => Not calculated
IWOHP => Not calculated
IWOBI => Not calculated
Population => Not calculated
Wannier interaction 2[1, 0, 0] <=> 1[0, 0, 0]
=============================================
DOS matrix => Not calculated
H_ij => Not calculated
P_ij => Not calculated
IWOHP => Not calculated
IWOBI => Not calculated
Population => Not calculated
Wannier interaction 2[1, 0, 0] <=> 3[0, 0, 0]
=============================================
DOS matrix => Not calculated
H_ij => Not calculated
P_ij => Not calculated
IWOHP => Not calculated
IWOBI => Not calculated
Population => Not calculated
Wannier interaction 2[1, 0, 0] <=> 4[0, 0, 0]
=============================================
DOS matrix => Not calculated
H_ij => Not calculated
P_ij => Not calculated
IWOHP => Not calculated
IWOBI => Not calculated
Population => Not calculated
Wannier interaction 2[1, 0, 0] <=> 7[0, 0, 0]
=============================================
DOS matrix => Not calculated
H_ij => Not calculated
P_ij => Not calculated
IWOHP => Not calculated
IWOBI => Not calculated
Population => Not calculated
Wannier interaction 5[1, 1, 1] <=> 1[0, 0, 0]
=============================================
DOS matrix => Not calculated
H_ij => Not calculated
P_ij => Not calculated
IWOHP => Not calculated
IWOBI => Not calculated
Population => Not calculated
Wannier interaction 5[1, 1, 1] <=> 3[0, 0, 0]
=============================================
DOS matrix => Not calculated
H_ij => Not calculated
P_ij => Not calculated
IWOHP => Not calculated
IWOBI => Not calculated
Population => Not calculated
Wannier interaction 5[1, 1, 1] <=> 4[0, 0, 0]
=============================================
DOS matrix => Not calculated
H_ij => Not calculated
P_ij => Not calculated
IWOHP => Not calculated
IWOBI => Not calculated
Population => Not calculated
Wannier interaction 5[1, 1, 1] <=> 7[0, 0, 0]
=============================================
DOS matrix => Not calculated
H_ij => Not calculated
P_ij => Not calculated
IWOHP => Not calculated
IWOBI => Not calculated
Population => Not calculated
Wannier interaction 6[0, 1, 0] <=> 1[0, 0, 0]
=============================================
DOS matrix => Not calculated
H_ij => Not calculated
P_ij => Not calculated
IWOHP => Not calculated
IWOBI => Not calculated
Population => Not calculated
Wannier interaction 6[0, 1, 0] <=> 3[0, 0, 0]
=============================================
DOS matrix => Not calculated
H_ij => Not calculated
P_ij => Not calculated
IWOHP => Not calculated
IWOBI => Not calculated
Population => Not calculated
Wannier interaction 6[0, 1, 0] <=> 4[0, 0, 0]
=============================================
DOS matrix => Not calculated
H_ij => Not calculated
P_ij => Not calculated
IWOHP => Not calculated
IWOBI => Not calculated
Population => Not calculated
Wannier interaction 6[0, 1, 0] <=> 7[0, 0, 0]
=============================================
DOS matrix => Not calculated
H_ij => Not calculated
P_ij => Not calculated
IWOHP => Not calculated
IWOBI => Not calculated
Population => Not calculated
As shown above, just as we can iterate over an AtomicInteractionContainer to yield individual AtomicInteraction objects, we can also iterate over an AtomicInteraction to yield a series of WannierInteraction objects, each of which represents the interaction between one of the Wannier functions associated with C8 and one of the Wannier functions associated with C9.
We can also use numpy-style indexing to pick out specific WannierInteraction objects, much like we can pick out individual AtomicInteraction objects from an AtomicInteractionContainer:
print(interactions[8, 9][0, 1])
Wannier interaction 0[0, 0, 1] <=> 1[0, 0, 0]
=============================================
DOS matrix => Not calculated
H_ij => Not calculated
P_ij => Not calculated
IWOHP => Not calculated
IWOBI => Not calculated
Population => Not calculated
If we provide only 1 index rather than 2, we can access all interactions between a given Wannier function and any other Wannier function:
for w_interaction in interactions[8, 9][0]:
print(w_interaction)
Wannier interaction 0[0, 0, 1] <=> 1[0, 0, 0]
=============================================
DOS matrix => Not calculated
H_ij => Not calculated
P_ij => Not calculated
IWOHP => Not calculated
IWOBI => Not calculated
Population => Not calculated
Wannier interaction 0[0, 0, 1] <=> 3[0, 0, 0]
=============================================
DOS matrix => Not calculated
H_ij => Not calculated
P_ij => Not calculated
IWOHP => Not calculated
IWOBI => Not calculated
Population => Not calculated
Wannier interaction 0[0, 0, 1] <=> 4[0, 0, 0]
=============================================
DOS matrix => Not calculated
H_ij => Not calculated
P_ij => Not calculated
IWOHP => Not calculated
IWOBI => Not calculated
Population => Not calculated
Wannier interaction 0[0, 0, 1] <=> 7[0, 0, 0]
=============================================
DOS matrix => Not calculated
H_ij => Not calculated
P_ij => Not calculated
IWOHP => Not calculated
IWOBI => Not calculated
Population => Not calculated
Note
If an AtomicInteractionContainer contains more than one interaction, the same logic can be applied to find all of the interactions between atom i and any other atom.
Now that we understand how to access individual WannierInteraction objects, let us proceed as we did in the basics and calculate the WOHP and the WOBI for the C-C bond between C8 and C9:
from pengwann.descriptors import DescriptorCalculator
from pengwann.io import read
from pengwann.occupations import get_occupation_matrix
kpoints, eigenvalues, u_matrices, hamiltonian = read(seedname='wannier90',
path='inputs')
mu = 10.5
nspin = 2
occupation_matrix = get_occupation_matrix(eigenvalues, mu, nspin)
num_wann = 8
energy_range = (-15, 26)
resolution = 0.1
sigma = 0.2
dcalc = DescriptorCalculator.from_eigenvalues(eigenvalues,
num_wann,
nspin,
energy_range,
resolution,
sigma,
kpoints,
u_matrices,
h=hamiltonian,
occupation_matrix=occupation_matrix)
interactions = dcalc.assign_descriptors(interactions)
Just like last time, we call the with_integrals() method to obtain the IWOHP and the IWOBI, but this time we add a new keyword argument:
interactions = interactions.with_integrals(dcalc.energies,
mu,
resolve_orbitals=True)
By setting resolve_orbitals = True, we integrate not only the total WOHP and WOBI for the C-C bond, but also the individual WOHPs and WOBIs associated with each individual WannierInteraction. For example, picking out one specific interaction:
print(interactions[8, 9][0, 1])
Wannier interaction 0[0, 0, 1] <=> 1[0, 0, 0]
=============================================
DOS matrix => Calculated
H_ij => -3.564015
P_ij => 0.2635638912672347
IWOHP => Calculated
IWOBI => Calculated
Population => Calculated
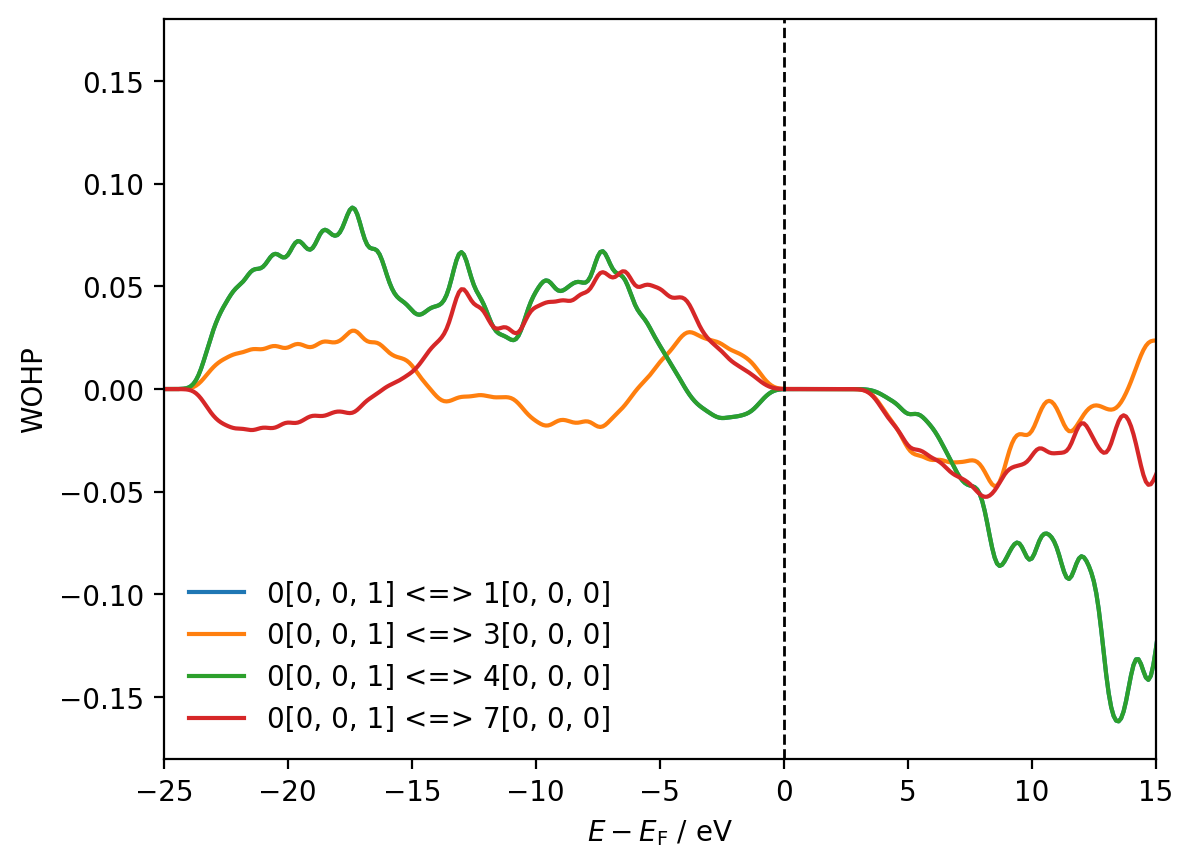
We are now able to access and plot WOHPs, WOBIs and their integrals for a series of individual WannierInteraction objects. For example, plotting all WOHPs and WOBIs for the interactions between Wannier function 0 and Wannier functions 1, 3, 4 and 7:
import matplotlib.pyplot as plt
shifted_energies = dcalc.energies - mu
fig, ax = plt.subplots()
for w_interaction in interactions[8, 9][0]:
ax.plot(shifted_energies, w_interaction.wohp, label=f"{w_interaction.tag}")
print(f"{w_interaction.tag} IWOHP = {w_interaction.iwohp:.2f}")
ax.axvline(x=0, color='black', ls='--', lw=1)
ax.set_xlim(-25, 15)
ax.set_ylim(-0.18, 0.18)
ax.set_xlabel(r'$E - E_{\mathrm{F}}$ / eV')
ax.set_ylabel('WOHP')
ax.legend(frameon=False)
plt.show()
0[0, 0, 1] <=> 1[0, 0, 0] IWOHP = 0.94
0[0, 0, 1] <=> 3[0, 0, 0] IWOHP = 0.18
0[0, 0, 1] <=> 4[0, 0, 0] IWOHP = 0.94
0[0, 0, 1] <=> 7[0, 0, 0] IWOHP = 0.41
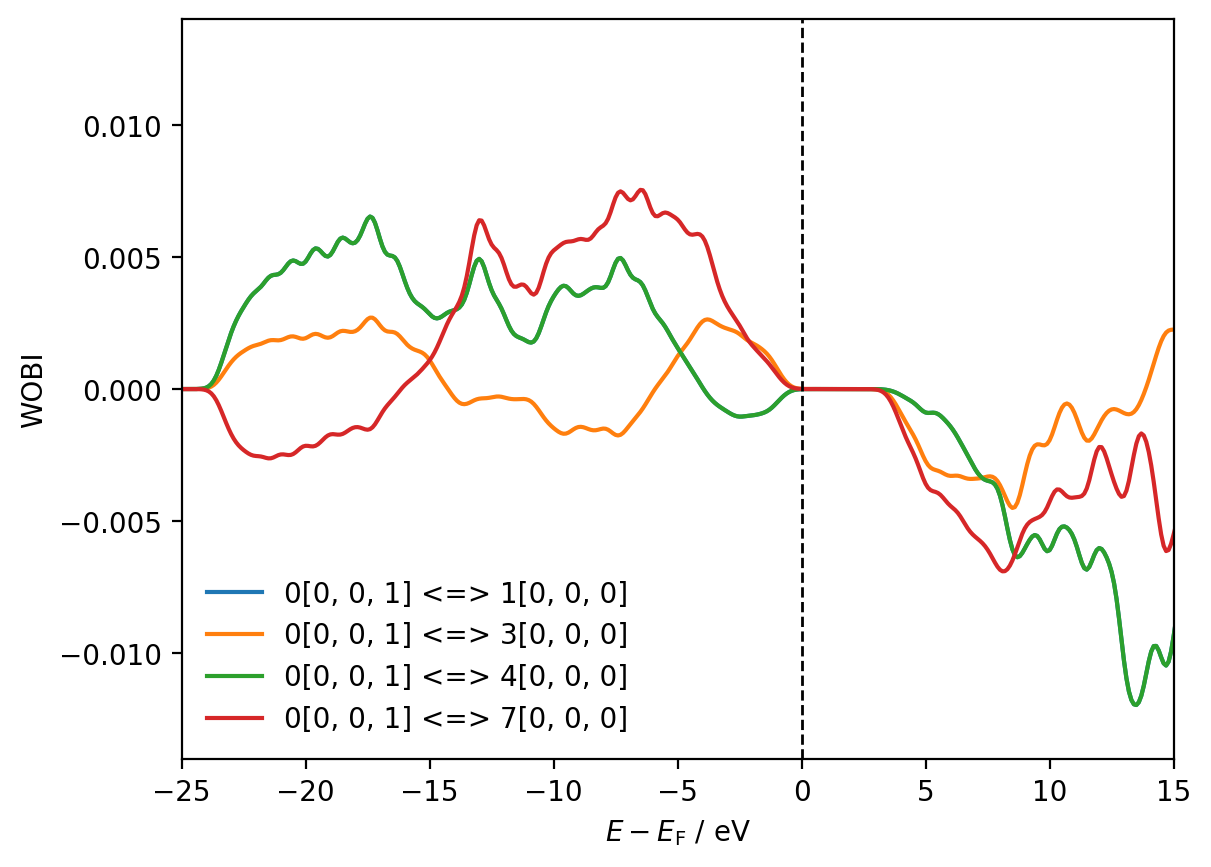
fig, ax = plt.subplots()
for w_interaction in interactions[8, 9][0]:
ax.plot(shifted_energies, w_interaction.wobi, label=f"{w_interaction.tag}")
print(f"{w_interaction.tag} IWOBI = {w_interaction.iwobi:.2f}")
ax.axvline(x=0, color='black', ls='--', lw=1)
ax.set_xlim(-25, 15)
ax.set_ylim(-0.014, 0.014)
ax.set_xlabel(r'$E - E_{\mathrm{F}}$ / eV')
ax.set_ylabel('WOBI')
ax.legend(frameon=False)
plt.show()
0[0, 0, 1] <=> 1[0, 0, 0] IWOBI = 0.07
0[0, 0, 1] <=> 3[0, 0, 0] IWOBI = 0.02
0[0, 0, 1] <=> 4[0, 0, 0] IWOBI = 0.07
0[0, 0, 1] <=> 7[0, 0, 0] IWOBI = 0.05
Note
The WOHP and the WOBI arising from the interaction between Wannier function 0[0, 0, 1] and Wannier function 1[0, 0, 0] (blue line) are not visible in the plots above because they are overlapped almost exactly by the WOHP and the WOBI arising from the interaction between Wannier function 0[0, 0, 1] and Wannier function 4[0, 0, 0].
k-resolved descriptors#
As well as resolving bonding descriptors with respect to individual Wannier functions, pengwann also facilitates the calculation of k-resolved descriptors:
interactions = dcalc.assign_descriptors(interactions, resolve_k=True)
print(interactions[8, 9].wohp.shape)
(411, 4096)
As seen above, the total WOHP for the interaction between C8 and C9 now has shape (411, 4096), which reflects its dimensions of \(N_{\mathrm{energies}} \times N_{\mathrm{kpoints}}\):
print(len(dcalc.energies))
print(len(kpoints))
411
4096
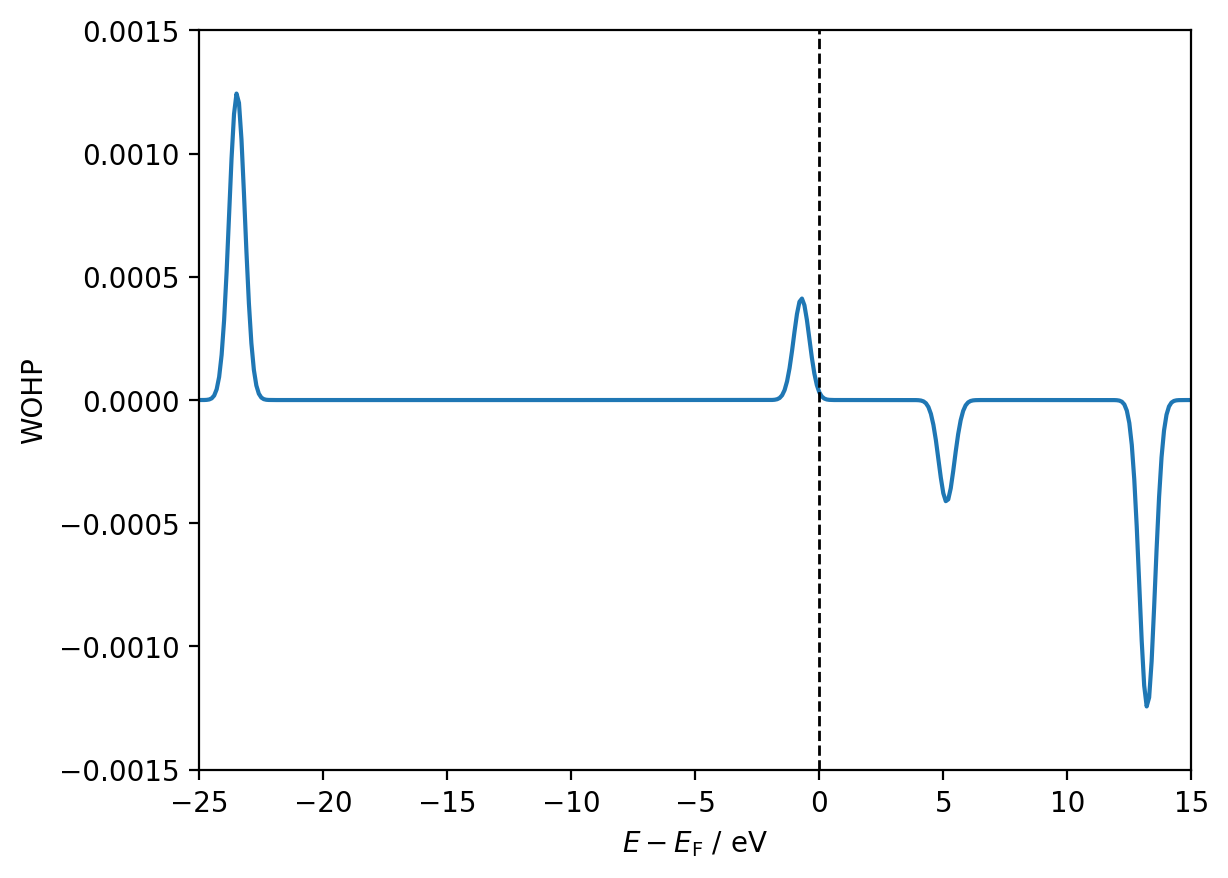
We can therefore access the WOHP for a specific k-point by indexing for a particular column. For example, the first k-point in the k-point mesh (for this particular example) is the Gamma point at (0, 0, 0) in the Brillouin zone:
fig, ax = plt.subplots()
gamma_wohp = interactions[8, 9].wohp[:, 0]
ax.plot(shifted_energies, gamma_wohp)
ax.axvline(x=0, color='black', ls='--', lw=1)
ax.set_xlim(-25, 15)
ax.set_ylim(-0.0015, 0.0015)
ax.set_xlabel(r'$E - E_{\mathrm{F}}$ / eV')
ax.set_ylabel('WOHP')
plt.show()
We can calculate the k-resolved IWOHP in exactly the same way we would for the non-k-resolved case:
interactions = interactions.with_integrals(dcalc.energies, mu)
print(interactions[8, 9].iwohp.shape)
(4096,)
As indicated above, the iwohp attribute is now an array of \(N_{\mathrm{kpoints}}\) values, so we can access the IWOHP for any individual k-point:
print(f"IWOHP at the Gamma point = {interactions[8, 9].iwohp[0]:.4f}")
IWOHP at the Gamma point = 0.0013